This Alnylam promotional website is intended for UK Healthcare Professionals Only. Adverse Event Reporting information can be found at the bottom of the page. Prescribing Information links can be found with the content that they relate to.



Primary Hyperoxaluria Type 1 (PH1)
Primary Hyperoxaluria Type 1 (PH1)
Primary Hyperoxaluria Type 1 (PH1) is a rare metabolic disorder caused by mutations in the alanine glyoxylate aminotransferase (AGXT) gene that result in a deficiency of liver-specific peroxisomal alanine glyoxylate aminotransferase (AGT) and consequent overproduction of oxalate by the liver. When the kidneys can no longer clear excess oxalate, calcium oxalate (CaOx) crystals deposit in other tissues, leading to systemic oxalosis.1
Resources


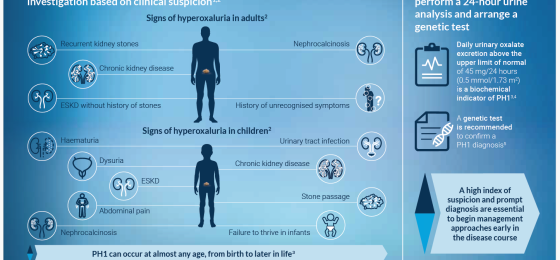
Therapies
OXLUMO® (lumasiran)
Explore clinical and educational resources related to the use of lumasiran in patients in all age groups with primary hyperoxaluria type 1 (PH1).
Congresses

Primary Hyperoxaluria Type 1 (PH1)
American Society of Nephrology (ASN) 2025

Primary Hyperoxaluria Type 1 (PH1)
European Society for Paediatric Nephrology (ESPN) 2025

Primary Hyperoxaluria Type 1 (PH1)
International Paediatric Transplant Association (IPTA) 2025
Publications
Primary Hyperoxaluria Type 1 (PH1)
Efficacy and Safety of Lumasiran for Advanced Primary Hyperoxaluria Type 1: 24-Month Follow-Up of the Phase 3 Illuminate-C Trial
American Journal of Kidney Diseases
References:
1. Fargue S, Acquaviva Bourdain C. Clin Kidney J. 2022;15(1):i4-i8.

Preparation Date: January 2026 Job Code: MB-UK-00039